Research
Materials chemistry is at a critical juncture with a massive growth of data from computational and experimental sources, made possible by efficient ab-initio methods, rapidly expanding computing resources, as well as recent advances in high-throughput/robotic laboratories. These data repositories now provide ready access to millions of datapoints of materials properties for use in chemical and materials informatics. Such resources have the potential to greatly accelerate scientific knowledge and engineering, provided suitable methods exist to effectively and reliably leverage the data. However, this can be challenging due to the need to account for the many intrinsic physical (quantum mechanical) complexities underlying these systems, necessitating the careful application of machine learning models and the effective inclusion of domain information and physical constraints. Our group at Georgia Tech develops novel computational approaches which can capture these complexities for use in mapping or populating the chemical/materials space. These include data-driven methods for learning representations of atomistic systems, fitting potential energy surfaces, obtaining interpretable latent spaces of chemical systems, and for building new materials at the atomic level.
We are interested in applying these methods to discover and design new functional materials for applications including catalysis, carbon capture and utilization, energy storage, and neuromorphic devices. We also seek to understand the underlying physiochemical phenomena governing the properties of materials from a data-driven perspective, relating to molecular activation on surfaces, chemical bonding in solids and interfaces, and strong electron correlation. Some specific ongoing projects are as follows:
Surrogate models for quantum chemistry
Ab initio quantum chemistry simulations are widely employed to obtain a broad range of materials properties, and its usage takes up almost a third of the share of computational resources on current high-performance computing centers. The cost of these calculations, especially at the higher levels of theory needed to provide accurate energetics, model excited states, and capture strong electron correlation represents a substantial bottleneck for computational studies and makes the study of these complex phenomena prohibitively expensive. To address this problem, we develop machine learning models which act as surrogates to these quantum chemistry methods for systems of arbitrary structural and compositional complexity and scalable to large system sizes.
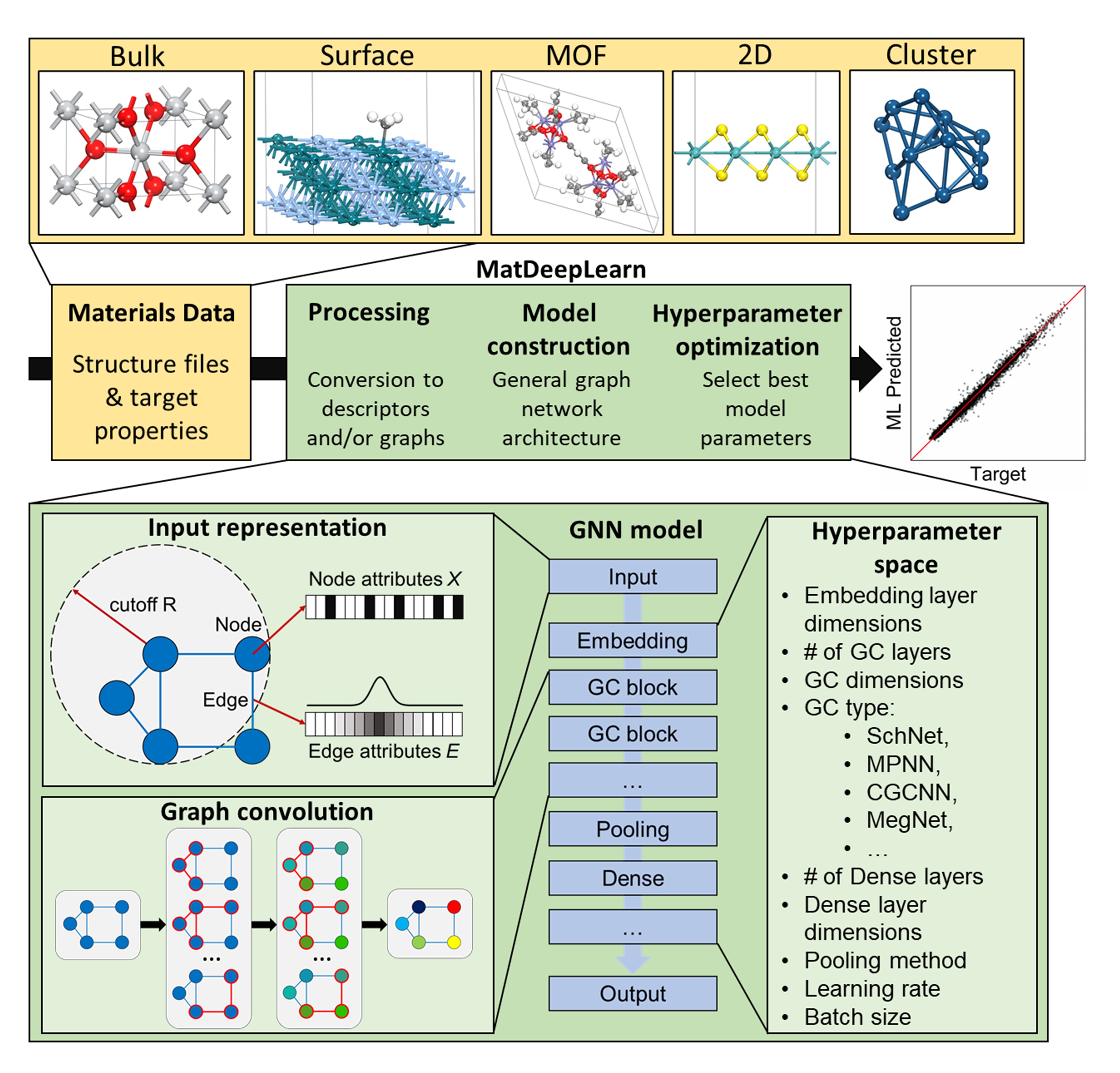
We are currently developing graph neural network models which takes atomic structure as input and predicts energies, forces, as well as a wide range of other materials properties such as density of states.
Data-driven inverse design
One of the greatest challenges in chemical/materials discovery is being able to design completely novel materials given a set of desired properties or functionalities, or inverse design. Traditionally, this can be a long and arduous process requiring human (chemical) intuition and existing design principles to accomplish. However, human-based design can barely keep up with the rapidly expanding repositories of scientific data available for query, nor can it satisfy the needs of high throughput synthesis and automated experimentation workflows. Here we investigate ways to accelerate or automate this design process through computational methods and data-driven approaches such as generative models. Examples include using invertible neural networks to solve inverse problems in materials design, and creating algorithms for invertible representations for use in atomic structure generation.
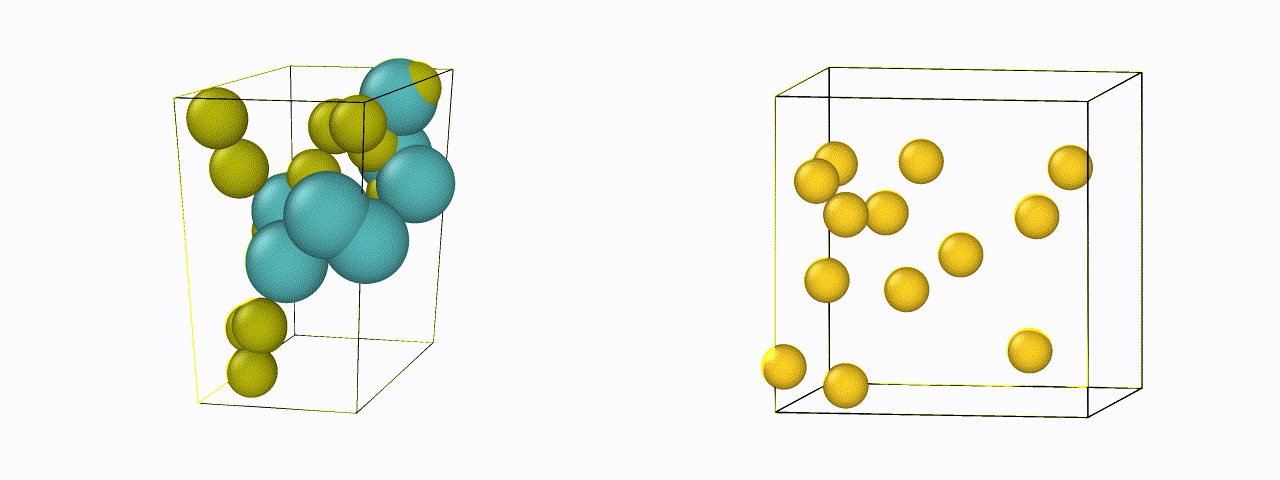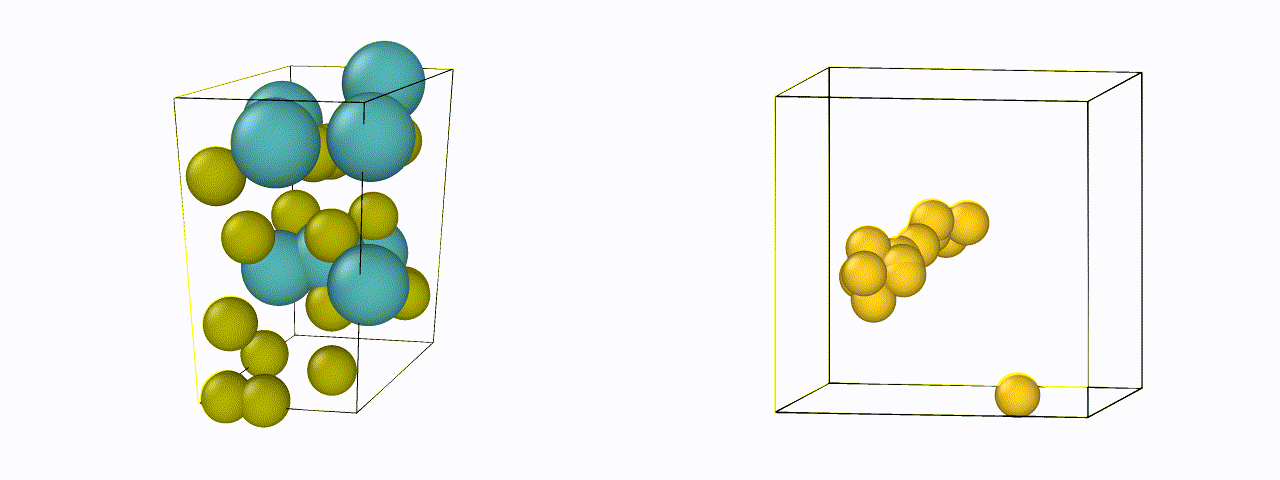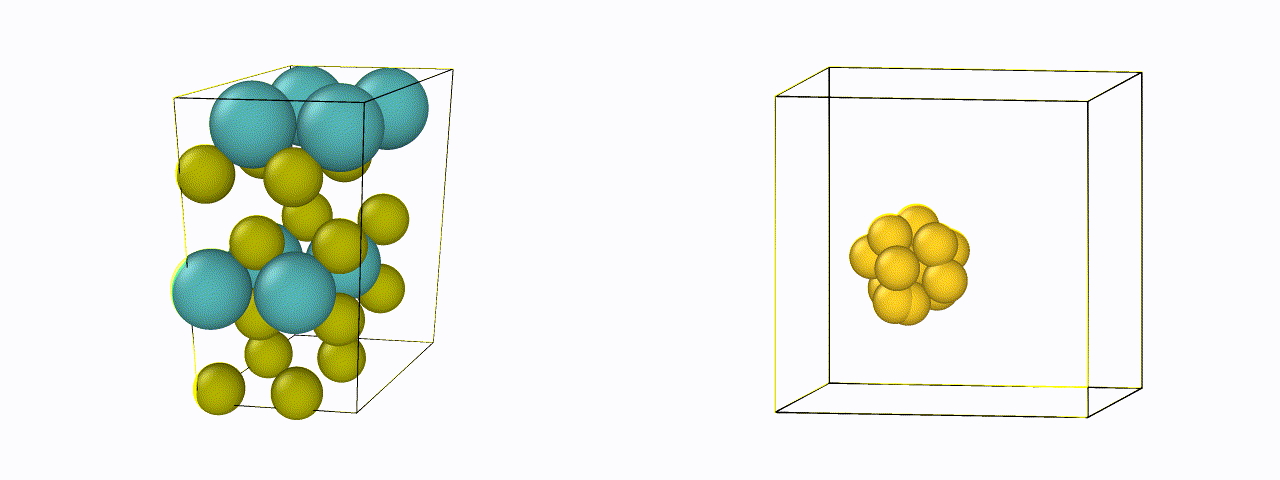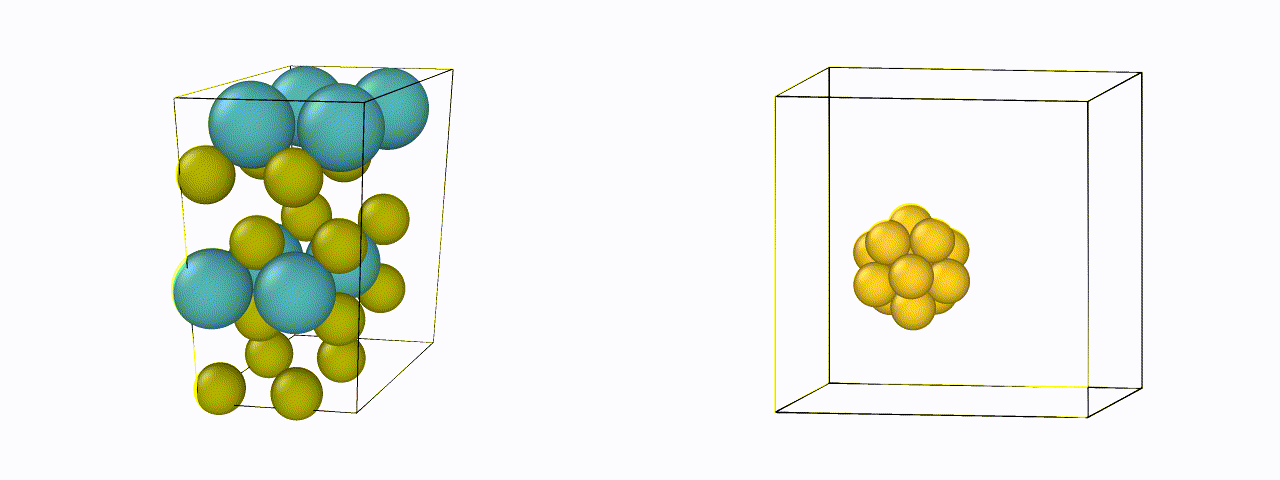
To design a new material, just defining the chemical compositions are not enough - it is also necessary to know their atomic or crystal structure. For efficient generation, an invertible representation of the atomic structure is needed which satisfies rotational, translational, and permutational invariances. We are currently exploring new representations of materials suitable for structure generation, and designing algorithms to make existing representations invertible back to their positions.
Chemically-informed machine learning
Incorporating domain knowledge and ensuring physically meaningful or consistent results from the machine learning models can be a necessity in situations with small datasets, modelling non-equilibirum systems, and for problems which are extrapolative in nature. We explore how chemical knowledge and contraints can be used to improve the performance of machine learning models through various avenues. These include the engineering of chemically meaningful features, applying constraints to the model architechture, or by conditioning the training of the model itself to more meaningful solutions.
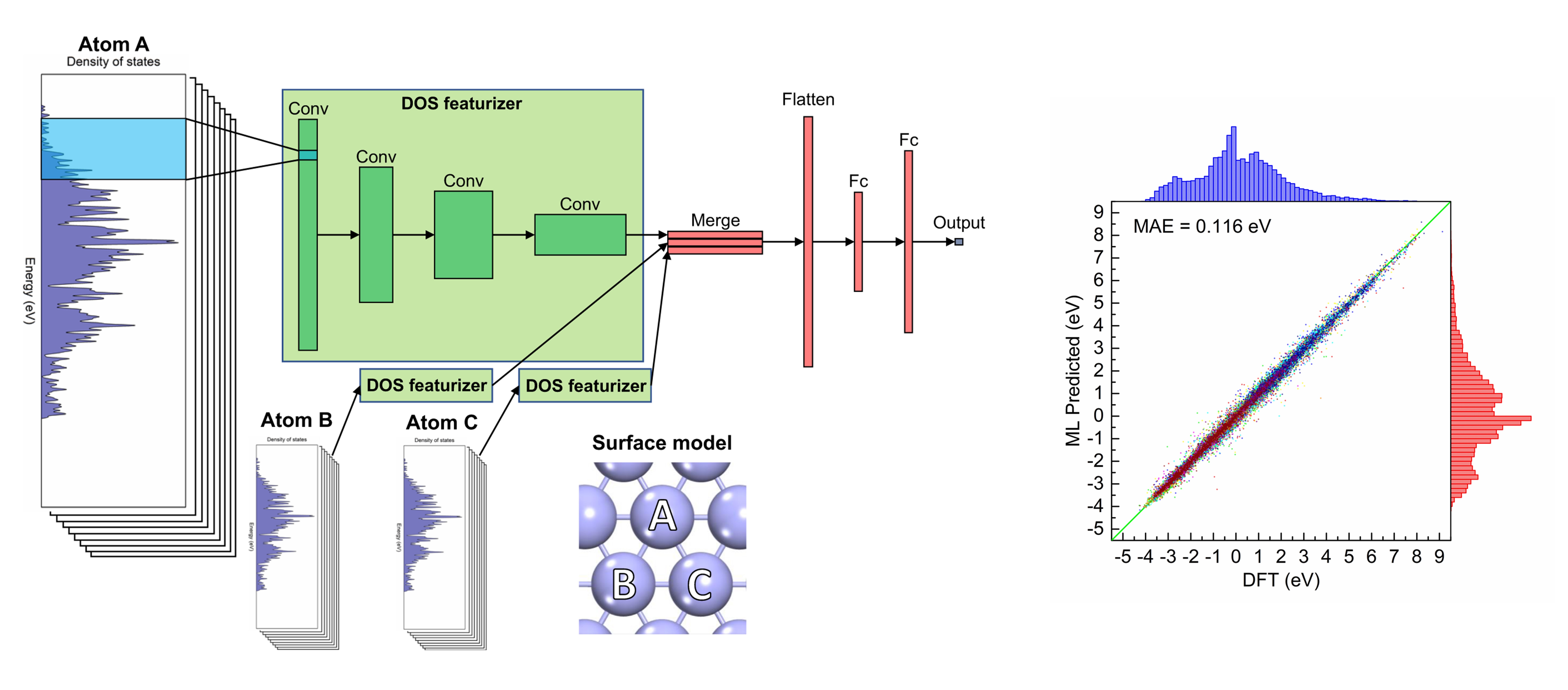
The density of states of a surface can reveal important information about their chemical properties, including its interaction with molecular adsorbates. We created a machine learning model using convolutional neural networks which can identify the features which govern adsorption and accurately predict the energetics of this process.
High-throughput computational simulations
In scientific machine learning, data can hardly be considered to be static but is instead constantly changing to reflect deficiencies in performance and changing problem specifications. Our group also performs quantum chemistry simulations, primarily density functional theory, in a high-throughput manner to provide data for machine learning models. We are working to close the loop between data generation and model development through automated simulation workflows and data sampling strategies.

We run our simulations on high performance supercomputers such as Perlmutter and Frontier.